Blutdirekt-PCR-Kit
Merkmale
■ Einfach und schnell: Die PCR-Amplifikation kann direkt mit Blut als Matrize durchgeführt werden, ohne die langwierigen Schritte der Probenvorbereitung und DNA-Extraktion.
■ Hohe Reinheit: Das Überspringen von Probenvorbehandlungs- und DNA-Extraktionsschritten kann helfen, Kreuzkontaminationen von Proben zu vermeiden.
■ Hoher Durchsatz: Die PCR-Identifikation für groß angelegte Proben kann durch die Kombination des Kits mit 96/384-Well-PCR-Platten durchgeführt werden.
■ Starke Universalität: Dieses Kit kann Fragmente mit hohem GC oder Fragmente mit komplexer Sekundärstruktur effizient amplifizieren und die Amplifikationslänge kann bis zu 5 kb betragen.
■ Starke Stressresistenz: Dieses Kit kann für verschiedene Tierarten und auf unterschiedliche Weise konservierte Blutproben verwendet werden.
Anwendungen
Die PCR-Produkte dieses Kits enthalten „A“ am 3'-Ende, das direkt für die Klonierung von TA-Vektoren verwendet werden kann. Dieses Kit kann für die Amplifikation von genomischen DNA-Fragmenten, genetische Hochdurchsatzanalysen und Genotypisierungsanalysen (z. B. Gennachweis) verwendet werden.
Alle Produkte können für ODM/OEM angepasst werden. Für Details,Bitte klicken Sie auf kundenspezifischen Service (ODM/OEM)




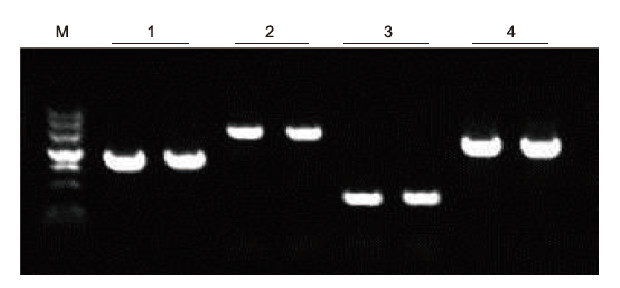 |
Unter Verwendung von humaner EDTA-Antikoagulation als Matrize wurden 4 Gene mit unterschiedlichen GC-Gehalten mit dem Blood Direct PCR Kit amplifiziert. Das PCR-Reaktionssystem war 20 µl, als Matrize wurde 1 µl Blut verwendet. M: TIANGEN-Marker II; 1: Fragmentgröße 1090 bp, GC-Gehalt 68,1 %; 2: Fragmentgröße 1915 bp, GC-Gehalt 70,4%; 3: Fragmentgröße 448 bp, GC-Gehalt 74,8%; 4: Fragmentgröße 1527 bp, GC-Gehalt 61,5%. Experimentelle Ergebnisse: Das Blood Direct PCR Kit kann DNA-Fragmente mit einem GC-Gehalt im Bereich von 61,5%-74,8% effektiv amplifizieren, was darauf hindeutet, dass es in der Lage ist, GC-Fragmente zu amplifizieren. |
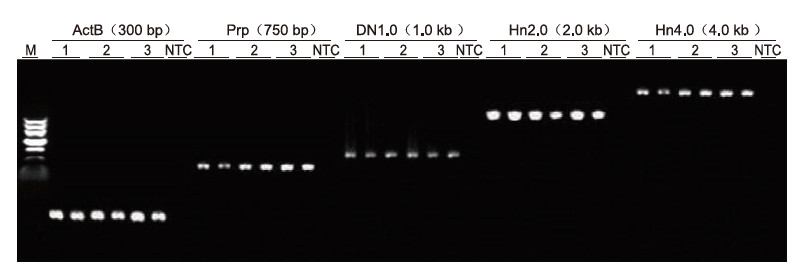 |
Unter Verwendung von humaner EDTA-Antikoagulation als Matrize wurden 5 Gene mit unterschiedlichen Längen (ActB, Prp, DN1.0, Hn2.0 und Hn4.0) mit dem Blood Direct PCR Kit amplifiziert. Das PCR-Reaktionssystem war 20 µl, als Matrize wurde 1 µl Blut verwendet. M: TIANGEN-Marker II; 1-3: 3 verschiedene Blutproben; NTC: Kontrolle ohne Primer. Experimentelle Ergebnisse: Das Blood Direct PCR Kit kann Fragmente mit einer Länge von bis zu 4 kb amplifizieren, was darauf hindeutet, dass es in der Lage ist, lange Fragmente zu amplifizieren. |
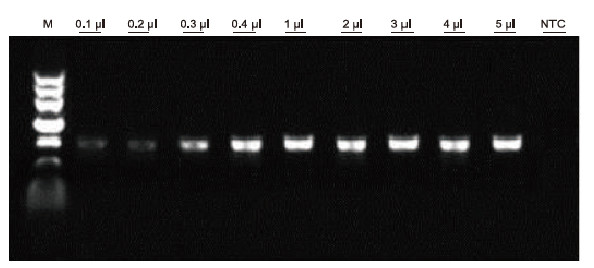 |
Unter Verwendung von humaner EDTA-Antikoagulation als Matrize wurde das Blood Direct PCR Kit für den PCR-Nachweis verschiedener Blutproben verwendet. Das PCR-Reaktionssystem war 20 µl, als Matrize wurde 1 µl Blut verwendet. M: TIANGEN-Marker II; 1-9: die Beladungsmenge an Blut beträgt 0,1 µl, 0,2 µl, 0,3 µl, 0,4 µl, 1 µl, 2 µl, 3 µl, 4 µl bzw. 5 µl; NTC: Steuerung ohne Vorlage Experimentelle Ergebnisse: Das Blood Direct PCR Kit weist eine starke Beständigkeit gegen Blut auf und kann Blutproben mit einem Beladungsbereich von 0,1-5 μl amplifizieren. |
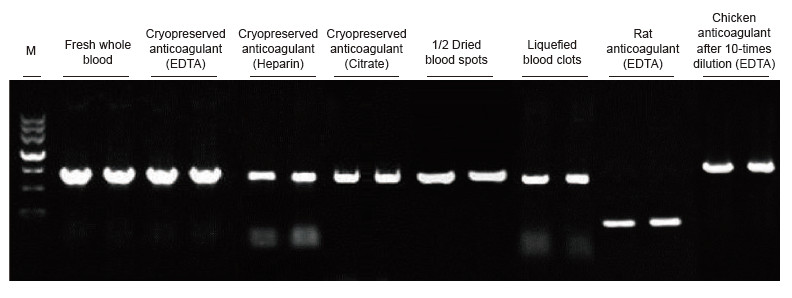 |
Als Vorlagen wurden Blutproben von Mensch, Ratte, Huhn und anderen Spezies mit unterschiedlichen Behandlungen verwendet. Der Blood Direct PCR Kit wurde verwendet, um PRNP (Mensch, 750 bp), Actin (Ratte, 200 bp) und β-Actin (Huhn, 1,0 kb) zu amplifizieren. Das PCR-Reaktionssystem war 20 µl, als Matrize wurde 1 µl Blut verwendet. M: TIANGEN-Marker II. Experimentelle Ergebnisse: Das Blood Direct PCR Kit kann auf eine Vielzahl von Proben angewendet werden, und der direkte PCR-Nachweis kann an Blutproben verschiedener Spezies mit unterschiedlichen Behandlungen durchgeführt werden. |
A-1 Vorlage
■ Das Template enthält Proteinverunreinigungen oder Taq-Inhibitoren usw. ——Reinigen Sie das DNA-Template, entfernen Sie Proteinverunreinigungen oder extrahieren Sie Template-DNA mit Reinigungskits.
■ Die Denaturierung des Templates ist nicht abgeschlossen ——Erhöhen Sie die Denaturierungstemperatur entsprechend und verlängern Sie die Denaturierungszeit.
■ Vorlagenverschlechterung – – Bereiten Sie die Vorlage erneut vor.
A-2 Grundierung
■ Schlechte Qualität der Grundierungen ——Resynthetisieren Sie die Grundierung.
■ Primerabbau ——Aliquotieren Sie die hochkonzentrierten Primer zur Konservierung in ein kleines Volumen. Mehrfaches Einfrieren und Auftauen oder Langzeit-Kryokonservierung bei 4 °C vermeiden.
■ Unsachgemäßes Design von Primern (zB Primerlänge nicht ausreichend, Dimer zwischen Primern gebildet usw.) -Redesign von Primern (Vermeidung der Bildung von Primer-Dimer und Sekundärstruktur)
A-3 mg2+Konzentration
■ Mg2+ Konzentration ist zu niedrig ——Mg . richtig erhöhen2+ Konzentration: Optimierung des Mg2+ Konzentration durch eine Reihe von Reaktionen von 1 mM bis 3 mM mit einem Intervall von 0,5 mM, um das optimale Mg . zu bestimmen2+ Konzentration für jede Matrize und jeden Primer.
A-4 Glühtemperatur
■ Die hohe Annealing-Temperatur beeinflusst die Bindung von Primer und Template. ——Reduzieren Sie die Glühtemperatur und optimieren Sie den Zustand mit einem Gradienten von 2°C.
A-5 Verlängerungszeit
■ Kurze Verlängerungszeit——Verlängert die Verlängerungszeit.
Phänomene: Negative Proben zeigen auch die Zielsequenzbanden.
A-1 Kontamination von PCR
■ Kreuzkontamination von Zielsequenz- oder Amplifikationsprodukten ——Die Probe, die die Zielsequenz enthält, vorsichtig in die negative Probe pipettieren oder aus dem Zentrifugenröhrchen verschütten. Die Reagenzien oder Geräte sollten autoklaviert werden, um vorhandene Nukleinsäuren zu eliminieren, und das Vorliegen einer Kontamination sollte durch Negativkontrollexperimente bestimmt werden.
■ Kontamination mit Reagenzien ——Aliquotieren Sie die Reagenzien und lagern Sie sie bei niedriger Temperatur.
A-2 Primer
■ Mg2+ Konzentration ist zu niedrig ——Mg . richtig erhöhen2+ Konzentration: Optimierung des Mg2+ Konzentration durch eine Reihe von Reaktionen von 1 mM bis 3 mM mit einem Intervall von 0,5 mM, um das optimale Mg . zu bestimmen2+ Konzentration für jede Matrize und jeden Primer.
■ Unsachgemäßes Primer-Design und die Zielsequenz weist Homologie mit der Nicht-Zielsequenz auf. ——Grundierungen neu gestalten.
Phänomene: Die PCR-Amplifikationsbanden stimmen nicht mit der erwarteten Größe überein, entweder groß oder klein, oder manchmal treten sowohl spezifische Amplifikationsbanden als auch unspezifische Amplifikationsbanden auf.
A-1 Grundierung
■ Schlechte Primer-Spezifität
——Grundierung neu gestalten.
■ Die Primerkonzentration ist zu hoch ——Erhöhen Sie die Denaturierungstemperatur richtig und verlängern Sie die Denaturierungszeit.
A-2 mg2+ Konzentration
■ Das Mg2+ Konzentration zu hoch ——Mg2+ Konzentration richtig reduzieren: Mg . optimieren2+ Konzentration durch eine Reihe von Reaktionen von 1 mM bis 3 mM mit einem Intervall von 0,5 mM, um das optimale Mg . zu bestimmen2+ Konzentration für jede Matrize und jeden Primer.
A-3 Thermostabile Polymerase
■ Übermäßige Enzymmenge ——Verringern Sie die Enzymmenge entsprechend in Intervallen von 0,5 U.
A-4 Glühtemperatur
■ Die Glühtemperatur ist zu niedrig ——Erhöhen Sie die Glühtemperatur entsprechend oder wenden Sie die zweistufige Glühmethode an
A-5 PCR-Zyklen
■ Zu viele PCR-Zyklen ——Reduzieren Sie die Anzahl der PCR-Zyklen.
A-1 Grundierung——Schlechte Spezifität ——Entwerfen Sie den Primer neu, ändern Sie die Position und Länge des Primers, um seine Spezifität zu verbessern; oder verschachtelte PCR durchführen.
A-2 Template-DNA
——Die Matrize ist nicht rein ——Reinigen Sie die Matrize oder extrahieren Sie DNA mit Aufreinigungskits.
A-3 mg2+ Konzentration
——Mg2+ Konzentration zu hoch ——Mg . richtig reduzieren2+ Konzentration: Optimierung des Mg2+ Konzentration durch eine Reihe von Reaktionen von 1 mM bis 3 mM mit einem Intervall von 0,5 mM, um das optimale Mg . zu bestimmen2+ Konzentration für jede Matrize und jeden Primer.
A-4 dNTP
——Die dNTP-Konzentration ist zu hoch ——Reduzieren Sie die dNTP-Konzentration entsprechend
A-5 Glühtemperatur
——Zu niedrige Glühtemperatur ——Glühtemperatur entsprechend erhöhen
A-6 Zyklen
——Zu viele Zyklen ——Optimieren Sie die Zyklenzahl

Der erste Schritt besteht darin, die geeignete Polymerase auszuwählen. Reguläre Taq-Polymerase kann aufgrund fehlender 3'-5'-Exonuklease-Aktivität nicht Korrektur gelesen werden, und eine Fehlpaarung verringert die Verlängerungseffizienz von Fragmenten stark. Daher kann normale Taq-Polymerase Zielfragmente, die größer als 5 kb sind, nicht effektiv amplifizieren. Taq-Polymerase mit spezieller Modifikation oder eine andere High-Fidelity-Polymerase sollte ausgewählt werden, um die Verlängerungseffizienz zu verbessern und die Anforderungen der Amplifikation langer Fragmente zu erfüllen. Außerdem erfordert die Amplifikation langer Fragmente auch eine entsprechende Anpassung von Primerdesign, Denaturierungszeit, Extensionszeit, Puffer-pH usw. Üblicherweise können Primer mit 18-24 bp zu einer besseren Ausbeute führen. Um Matrizenschäden zu vermeiden, sollte die Denaturierungszeit bei 94 °C auf 30 Sekunden oder weniger pro Zyklus reduziert werden und die Zeit zum Temperaturanstieg auf 94 °C vor der Amplifikation sollte weniger als 1 Minute betragen. Darüber hinaus kann eine effektive Amplifikation langer Fragmente durch Einstellen der Extensionstemperatur auf etwa 68 °C und Gestaltung der Extensionszeit entsprechend der Rate von 1 kb/min sichergestellt werden.
Die Fehlerrate der PCR-Amplifikation kann durch die Verwendung verschiedener DNA-Polymerasen mit hoher Genauigkeit reduziert werden. Unter allen bisher gefundenen Taq-DNA-Polymerasen hat das Pfu-Enzym die niedrigste Fehlerrate und die höchste Genauigkeit (siehe beigefügte Tabelle). Neben der Enzymauswahl können Forscher die PCR-Mutationsrate weiter reduzieren, indem sie die Reaktionsbedingungen optimieren, einschließlich der Optimierung der Pufferzusammensetzung, der Konzentration der thermostabilen Polymerase und der Optimierung der PCR-Zykluszahl.
Produktkategorien
WARUM UNS WÄHLEN
Seit seiner Gründung entwickelt unsere Fabrik erstklassige Produkte unter Einhaltung des Prinzips
der Qualität zuerst. Unsere Produkte haben einen ausgezeichneten Ruf in der Branche und ein wertvolles Vertrauen bei neuen und alten Kunden gewonnen.
- Tel: +86 010-59822688
- Gebäude 5, Nr. 86, Shuangying West Road, Bezirk Changping, Peking.
- people@tiangen.com



