TIANSeq Fragment-/Reparatur-/Tailing-Modul
Merkmale
■ Einfache Anwendung: Einstufiger Abschluss der doppelsträngigen DNA-Fragmentierung, Endreparatur, dA-Tailing-Reaktion.
■ Hohe Effizienz der Bibliotheksumwandlung: Die hocheffiziente Bibliothekskonstruktion kann für 1 ng DNA-Proben gewährleistet werden.
■ Kosteneffizient: Es werden keine speziellen Instrumente und Geräte benötigt.
Spezifikation
Typ: Fragmentierung, Endreparatur und 3'-Ende-dA-Tailing von doppelsträngiger DNA
Stichprobe: Genomische DNA oder große Fragment-DNA
Argument: doppelsträngige DNA
Beispieleingabe starten: 1 ng - 1 μg
Betriebszeit: 34-66 Minuten
Nachgelagerte Anwendungen: Adapterligation für die Vorbereitung von DNA-Sequenzierungsbibliotheken
Alle Produkte können für ODM/OEM angepasst werden. Für Details,Bitte klicken Sie auf kundenspezifischen Service (ODM/OEM)




Gegenwärtig basiert die Hochdurchsatz-Sequenzierungstechnologie hauptsächlich auf der Sequenzierungstechnologie der nächsten Generation. Da die Leselänge der Sequenzierungstechnologie der nächsten Generation begrenzt ist, müssen wir die vollständige Sequenz zur Sequenzierung in kleine Fragmentbibliotheken aufteilen. Je nach den Anforderungen verschiedener Sequenzierungsexperimente wählen wir normalerweise Single-Ended-Sequenzierung oder Double-Ended-Sequenzierung. Derzeit sind die DNA-Fragmente der Sequenzierungsbibliothek der nächsten Generation im Allgemeinen im Bereich von 200-800 bp verteilt.
a) DNA ist von schlechter Qualität und enthält Inhibitoren. Verwenden Sie hochwertige DNA-Proben, um eine Hemmung der Enzymaktivität zu vermeiden.
b) Die Menge der DNA-Probe ist unzureichend, wenn die PCR-freie Methode zum Aufbau der DNA-Bibliothek verwendet wird. Wenn der Input der fragmentierten DNA 50 ng überschreitet, kann während des Bibliotheksaufbaus selektiv ein PCR-freier Workflow durchgeführt werden. Wenn die Kopienzahl der Bibliothek zu gering ist, um direkt sequenziert zu werden, kann die DNA-Bibliothek nach der Adapterligation durch PCR amplifiziert werden.
c) RNA-Kontamination führt zu einer ungenauen anfänglichen DNA-Quantifizierung Beim Reinigungsprozess von genomischer DNA kann eine RNA-Kontamination vorliegen, die zu einer ungenauen DNA-Quantifizierung und einer unzureichenden DNA-Beladung während der Bibliothekskonstruktion führen kann. RNA kann durch Behandlung mit RNase entfernt werden.
A-1
a) Kleine Fragmente (60 bp-120 bp) erscheinen Kleine Fragmente sind normalerweise Adapterfragmente oder Dimere, die von Adaptern gebildet werden. Die Aufreinigung mit Agencourt AMPure XP Magnetbeads kann diese Adapterfragmente effektiv entfernen und die Sequenzierungsqualität sicherstellen.
b) Große Fragmente erscheinen in der Bibliothek nach der PCR-Amplifikation Die Größe des DNA-Fragments der Bibliothek nimmt um 120 bp zu, nachdem der Adapter ligiert wurde. Wenn das DNA-Fragment nach der Adapterligation um mehr als 120 bp zunimmt, kann dies durch eine abnormale Fragmentamplifikation oder eine übermäßige PCR-Amplifikation verursacht werden. Eine Reduzierung der Anzahl der PCR-Zyklen kann die Situation verhindern.
c) Abnorme Größe von Bibliotheks-DNA-Fragmenten nach Adapterligation Die Länge des Adapters in diesem Kit beträgt 60 bp. Wenn die beiden Enden des Fragments an die Adapter ligiert werden, erhöht sich die Länge nur um 120 bp. Wenn Sie einen anderen als den in diesem Kit enthaltenen Adapter verwenden, wenden Sie sich bitte an den Lieferanten, um relevante Informationen wie die Adapterlänge bereitzustellen. Bitte stellen Sie sicher, dass der Ablauf und die Durchführung des Experiments den im Handbuch beschriebenen Schritten folgen.
d) Abnormale DNA-Fragmentgröße vor der Adapterligation Der Grund für dieses Problem kann durch falsche Reaktionsbedingungen während der DNA-Fragmentierung verursacht werden. Für unterschiedliche DNA-Eingaben sollten unterschiedliche Reaktionszeiten verwendet werden. Wenn der DNA-Input mehr als 10 ng beträgt, empfehlen wir als Startzeit für die Optimierung die Reaktionszeit von 12 min zu wählen und die zu diesem Zeitpunkt produzierte Fragmentgröße liegt hauptsächlich im Bereich von 300-500 bp. Benutzer können die Länge der DNA-Fragmente für 2-4 min nach ihren eigenen Anforderungen erhöhen oder verringern, um die DNA-Fragmente auf die erforderliche Größe zu optimieren.
A-2
a) Fragmentierungszeit ist nicht optimiert Wenn die fragmentierte DNA zu klein oder zu groß ist, beachten Sie bitte die Richtlinien für die Fragmentierungszeitauswahl in der Anleitung zur Bestimmung der Reaktionszeit und verwenden Sie diesen Zeitpunkt als Kontrolle, zusätzlich einrichten a Reaktionssystem, um 3 Minuten zu verlängern oder zu verkürzen, um eine genauere Anpassung der Fragmentierungszeit vorzunehmen.
A-3
Abnorme Größenverteilung der DNA nach Fragmentierungsbehandlung
a) Falsche Auftaumethode des Fragmentierungsreagenzes oder das Reagenz wird nach dem Auftauen nicht vollständig gemischt. Tauen Sie das 5× Fragmentation Enzyme Mix-Reagenz auf Eis auf. Mischen Sie das Reagenz nach dem Auftauen gleichmäßig, indem Sie vorsichtig am Boden des Röhrchens klopfen. Reagenz nicht vortexen!
b) Die DNA-Eingabeprobe enthält EDTA oder andere Schadstoffe Die Abreicherung von Salzionen und Chelatbildnern im DNA-Reinigungsschritt ist für den Erfolg des Experiments besonders wichtig. Wenn DNA in 1 × TE gelöst ist, verwenden Sie die in der Anleitung beschriebene Methode, um die Fragmentierung durchzuführen. Wenn die EDTA-Konzentration in der Lösung unsicher ist, wird empfohlen, die DNA zu reinigen und für die anschließende Reaktion in entionisiertem Wasser aufzulösen.
c) Ungenaue anfängliche DNA-Quantifizierung Die Größe der fragmentierten DNA hängt eng mit der Menge der zugeführten DNA zusammen. Vor der Fragmentierungsbehandlung ist eine genaue Quantifizierung der DNA mit Qubit, Picogreen und anderen Methoden unerlässlich, um die genaue DNA-Menge im Reaktionssystem zu bestimmen.
d) Die Herstellung des Reaktionssystems erfolgt nicht nach Vorschrift Die Herstellung des fragmentierten Reaktionssystems muss streng nach Vorschrift auf Eis erfolgen. Um die beste Wirkung zu gewährleisten, sollten alle Reaktionskomponenten auf Eis gestellt werden und die Vorbereitung des Reaktionssystems sollte nach vollständiger Abkühlung erfolgen. Nachdem die Vorbereitung abgeschlossen ist, streichen oder pipettieren Sie, um gründlich zu mischen. Nicht wirbeln!
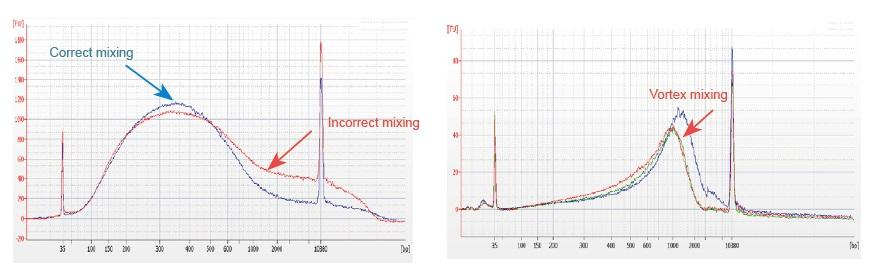
1. Eine unsachgemäße Mischmethode (Wirbel, heftige Schwingungen usw.) führt zu einer anormalen Verteilung der Bibliotheksfragmente (wie in der folgenden Abbildung gezeigt) und beeinträchtigt so die Qualität der Bibliothek. Daher beim Ansetzen der Fragmentation Mix-Reaktionslösung zum Mischen vorsichtig auf und ab pipettieren oder mit der Fingerspitze streichen und gleichmäßig mischen. Achten Sie darauf, sich nicht mit Vortex zu vermischen.
2. Für den Bibliotheksaufbau muss hochreine DNA verwendet werden
■ Gute DNA-Integrität: Die Elektrophorese-Bande beträgt mehr als 30 kb, ohne Tailing
■ OD260/230: >1,5
■ OD260/280: 1,7-1,9
3. Die DNA-Eingabemenge muss genau sein Es wird empfohlen, anstelle von Nanodrop die Methoden Qubit und PicoGreen zu verwenden, um die DNA zu quantifizieren.
4. Der EDTA-Gehalt in DNA-Lösung muss bestimmt werden EDTA hat einen großen Einfluss auf die Fragmentierungsreaktion. Bei hohem EDTA-Gehalt muss vor dem nachfolgenden Test eine DNA-Reinigung durchgeführt werden.
5. Die Fragmentierungsreaktionslösung muss auf Eis hergestellt werden Der Fragmentierungsprozess ist temperatur- und zeitabhängig (insbesondere nach Zugabe von Verstärker). Um die Genauigkeit der Reaktionszeit zu gewährleisten, bereiten Sie das Reaktionssystem bitte auf Eis vor.
6. Die Reaktionszeit der Fragmentierung muss genau sein Die Reaktionszeit des Fragmentierungsschritts beeinflusst direkt die Größe der Fragmentprodukte und beeinflusst somit die Größenverteilung der DNA-Fragmente in der Bibliothek.
1. Welcher Probentyp ist für dieses Kit geeignet?
Der geeignete Probentyp dieses Kits kann Gesamt-RNA oder gereinigte mRNA mit guter RNA-Integrität sein. Wenn Gesamt-RNA zum Aufbau der Bibliothek verwendet wird, wird empfohlen, zuerst das rRNA-Depletion-Kit (Kat.-Nr. 4992363/4992364/4992391) zu verwenden, um rRNA zu entfernen.
2. Können FFPE-Proben verwendet werden, um mit diesem Kit eine Bibliothek zu erstellen?
Die mRNA in FFPE-Proben wird bis zu einem gewissen Grad mit relativ schlechter Integrität abgebaut. Bei Verwendung dieses Kits für den Bibliotheksaufbau wird empfohlen, die Fragmentierungszeit zu optimieren (Fragmentierungszeit verkürzen oder keine Fragmentierung durchführen).
3. Wenn Sie den Schritt zur Größenauswahl im Produkthandbuch verwenden, was kann dazu führen, dass das eingefügte Segment eine leichte Abweichung aufweist?
Die Größenauswahl muss in strikter Übereinstimmung mit dem Schritt zur Größenauswahl in diesem Produkthandbuch erfolgen. Bei Abweichungen kann die Ursache darin liegen, dass die Magnetkügelchen nicht auf Raumtemperatur abgeglichen oder nicht vollständig gemischt sind, die Pipette nicht genau ist oder die Flüssigkeit in der Spitze zurückbleibt. Es wird empfohlen, für das Experiment die Spitzen mit geringer Adsorption zu verwenden.
4. Auswahl von Adaptern im Bibliotheksbau
Der Bibliotheksbausatz enthält kein Adapterreagenz und es wird empfohlen, diesen Kit zusammen mit dem TIANSeq Single-Index Adapter (Illumina) (4992641/4992642/4992378) zu verwenden.
5. QC der Bibliothek
Quantitativer Nachweis der Bibliothek: Qubit und qPCR werden verwendet, um die Massenkonzentration bzw. die molare Konzentration der Bibliothek zu bestimmen. Die Bedienung erfolgt ausschließlich in Übereinstimmung mit dem Produkthandbuch. Die Konzentration der Bibliothek wird im Allgemeinen die Anforderungen der NGS-Sequenzierung erfüllen. Erkennung des Verteilungsbereichs der Bibliothek: Verwendung des Agilent 2100 Bioanalyzer zur Erkennung des Verteilungsbereichs der Bibliothek.
6. Auswahl der Amplifikationszyklusnummer
Gemäß der Anleitung beträgt die Anzahl der PCR-Zyklen 6-12, und die Anzahl der benötigten PCR-Zyklen sollte entsprechend der Probeneingabe ausgewählt werden. In Bibliotheken mit hoher Ausbeute tritt in der Regel eine Überamplifikation in unterschiedlichem Ausmaß auf, was sich in einem etwas größeren Peak nach dem Peak des Zielbereichs beim Nachweis von Agilent 2100 Bioanalyzer manifestiert oder die nachgewiesene Konzentration von Qubit niedriger ist als die von qPCR. Eine leichte Überamplifikation ist ein normales Phänomen, das die Sequenzierung der Bibliothek und die anschließende Datenanalyse nicht beeinflusst.
7. Spikes erscheinen im Erkennungsprofil des Agilent 2100 Bioanalyzer
Das Auftreten von Spitzen bei der Detektion mit dem Agilent 2100 Bioanalyzer ist auf die ungleichmäßige Fragmentierung der Proben zurückzuführen, bei der mehr Fragmente in einer bestimmten Größe vorhanden sind, und dies wird nach der PCR-Anreicherung deutlicher. In diesem Fall wird empfohlen, die Größenauswahl nicht durchzuführen, dh die Fragmentierungsbedingung auf 94°C für 15 min inkubiert einzustellen, wo die Fragmentverteilung klein und konzentriert ist und die Homogenität verbessert werden kann.
Produktkategorien
WARUM UNS WÄHLEN
Seit seiner Gründung entwickelt unsere Fabrik erstklassige Produkte unter Einhaltung des Prinzips
der Qualität zuerst. Unsere Produkte haben einen ausgezeichneten Ruf in der Branche und ein wertvolles Vertrauen bei neuen und alten Kunden gewonnen.
- Tel: +86 010-59822688
- Gebäude 5, Nr. 86, Shuangying West Road, Bezirk Changping, Peking.
- people@tiangen.com



